
Post 52: The evolution of AlphaFold 🕦
Published:
AlphaFold (AF) is the most famous artificial intelligence (AI) in the scientific world because it was the first to solve the challenge of predicting the atomic structure of proteins, although not all interactions between biomolecules. However, the ideas behind each of its three versions are radically different.
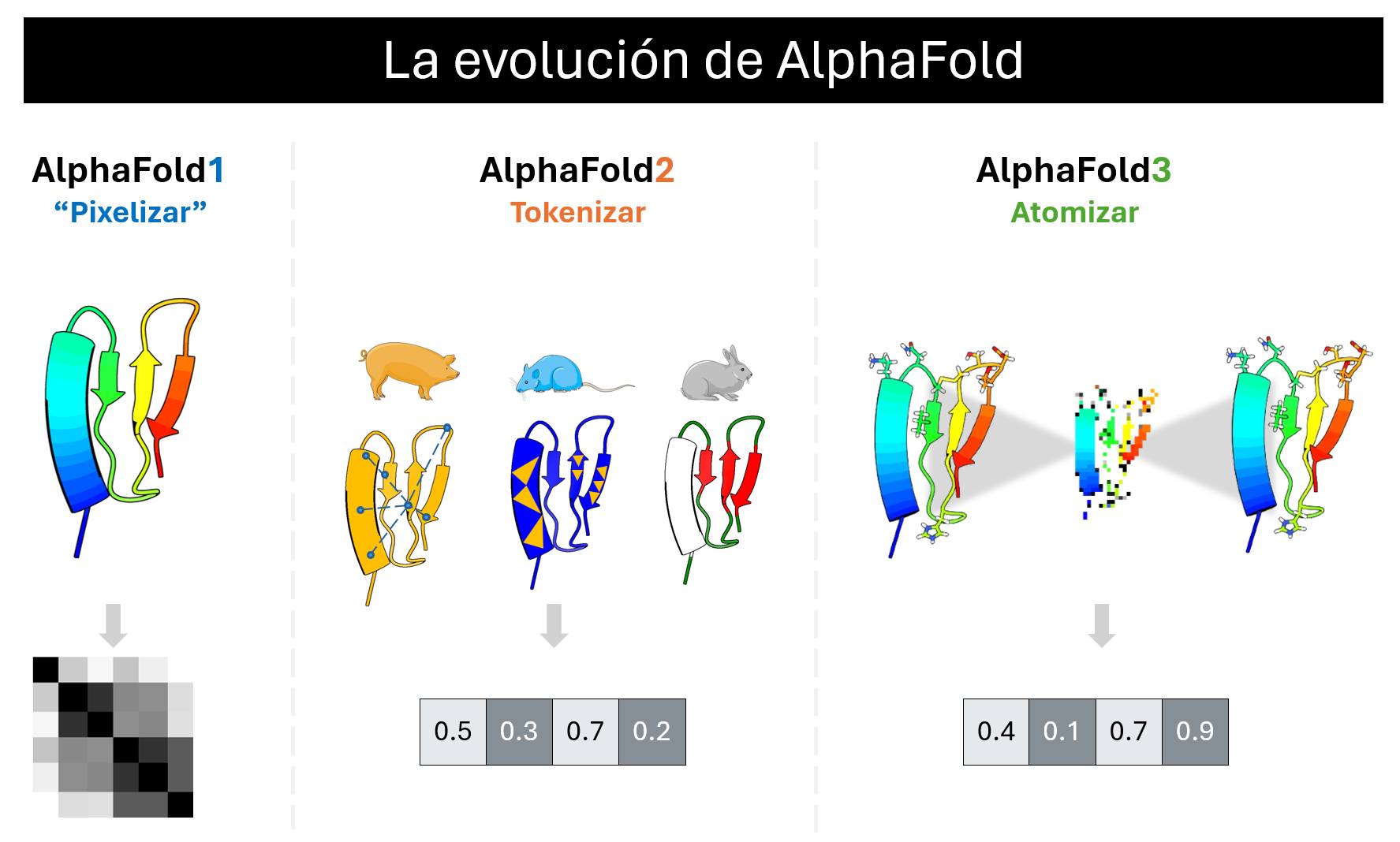
The innovation behind AF1 was to “pixelate” proteins, meaning to represent them as images that capture the distances between atoms. This way of representing proteins allowed them to be analyzed with an AI specialized in images, and although they did not manage to overcome the challenge, they outperformed all other competitors by a large margin.
In AF2, they realized that AF1 would not be enough, and it was necessary to discard everything and start anew. The innovation behind AF2 was to “tokenize” proteins, breaking them into pieces to allow an AI to autonomously learn to represent proteins as numerical vectors. This is very different, as in AF1 we guided the AI on how to represent proteins, but in AF2, the AI itself learned to represent them in the most optimal way possible. To enable AF2 to do this, it was necessary to write algorithms heavily inspired by the evolution, biochemistry, and biophysics of proteins, making it a highly valued algorithm among biologists.
However, AF2 fell victim to its own success. With so many bio-inspired bases in its algorithm, it did not generalize to the vast world of interactions between biomolecules. Thus, they had to discard everything and start over once again. The innovation behind AF3 was to “atomize” proteins, meaning they no longer just broke them into pieces but represented them as the smallest possible unit of molecules: the atom. By atomizing the algorithm, it was possible to discard practically all the biological bases that had been written for AF2, allowing the AI to learn these bases autonomously. Here again, proteins are represented as vectors, but now through a trial-and-error process where the AI reconstructs the atoms of the proteins.
In reality, none of AF’s innovations are truly original. All were previously explored ideas, but the scientists at Google managed to integrate them in a very thoughtful way so that, combined with their great computational power, they could train each version of AF. Moreover, it is quite surprising to see how most biological bases were discarded to increasingly favor the autonomy of the AI; which, while it serves quite well, seems to go against what we usually do in science: understanding something before moving on to its applications.
So, new ways of analyzing and/or approaching things open up new paths … sometimes quite interesting ones.
References:
- (On the paradigm shift from AF2 to AF3) The principle of uncertainty in biology: Will machine learning/artificial intelligence lead to the end of mechanistic studies?
- (On the transition from AF1 to AF2) Starting at Go: Protein structure prediction succumbs to machine learning
- (On the biological bases of AF2) Protein structure prediction by AlphaFold2: are attention and symmetries all you need?
And many other previous posts about AF:
- A very strange folding
- 200 Million new structures!
- A year after AlphaFold2
- How much did it cost to train AlphaFold2?
- AlphaFold2, the most famous scientific article
- How did AlphaFold2 learn to model proteins?
Addendum:
Want to know what existed before AlphaFold? Here’s a very comprehensive history of it from Quanta Magazine. Interesting are the quotes from several involved characters, such as Janet Thornton:
- Other AlphaFold competitors, including Meta, crafted their own algorithms […]. However […], no one has been able to match AlphaFold’s accuracy so far, Thornton said, “I’m sure they will, but I think getting another … AlphaFold moment like that will be very difficult.”
- How AI Revolutionized Protein Science
- Who is Janet Thornton?
AF2 is the “AlexNet” moment of the intersection between biology and AI. That is, the moment that convinced people to look at/use these tools. And it’s true enough that just look at the statistics of the article. Moreover, there is now a boom of bio-AI-generative startups (generatebiomedicines, profluent, DynoTx, Nabla Bio, etc.) as well as a considerable number of awards related to DeepMind or David Baker’s lab.
In particular, I am quite displeased with the generative nature of AF3 because it makes me distrust AF2. Furthermore, we should not confuse the CASP challenge (i.e., predicting the structure) with the dynamic behavior of proteins and their “folding” as an explanation of their structure-function.